


Introduction: Truncating mutations in Additional Sex Combs-Like 1 (ASXL1) are associated with a high-risk disease phenotype in myeloid malignancies. In chronic myelomonocytic leukemia (CMML), truncating ASXL1 mutations are known to increase transcriptional activity of leukemic driver genes and have been associated with gene body hypermethylation. We interrogated the transcriptome and methylome of patients with ASXL1-mutant (MT) and -wildtype (WT) CMML using a multi-omics approach to test the hypothesis that gene expression is mediated through gene body methylation.
Methods: Bone marrow mononuclear cells from patients with ASXL1 WT (n=8) and MT (n=8) CMML were subjected to targeted NGS of DNA, whole transcriptome shotgun sequencing (RNA-seq), and immunoprecipitation of DNA methyl residues (DIP-seq). After quality control all samples were sequenced on an Illumina HiSeq 4000 before further processing and data analysis. Differential gene expression analysis was performed to identify genes up-regulated in MT CMML. The samples in the two groups were treated as biological replicates and subjected to a consensus peak calling strategy requiring an overlap of at least 30% between samples and an adjusted p-value < 5x10-5 for a methylation peak to be considered statistically significant. For validation purposes methylation analysis was performed on 3 ASXL1 MT and 3 WT CMML patients using Illumina Infinium MethylationEPIC microarrays and differentially methylated regions were identified using a bump hunting strategy. Gene body methylation was defined as methylation in gene bodies (outside the promoter region, i.e. transcription start site ±2kb). Gene body methylation was compared between WT and MT CMML for the up-regulated genes and correlated with expression of all genes in MT CMML.
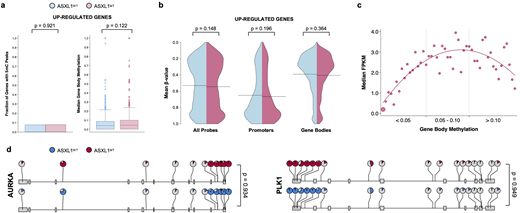
Results: Sixteen WHO-defined CMML patients were included, median age 69 years (48-77), 63% male, 50% had truncating ASXL1 frame shift mutations. Abnormal karyotypes were observed in the same number of patients and the burden of co-mutations was similar between the two groups (median number per group 3 vs. 3, p=0.508). This included several modulators of DNA methylation including TET2, DNMT3A, and IDH2 (median number per group 1 vs. 1, p=0.699). There was a predominant up-regulation of gene expression in MT CMML: 707 genes up- and 124 down-regulated (FDR<0.050) without evidence of differential methylation in promoter regions of the differentially expressed genes. There was no difference in the number of genes with consensus methylation peaks in the gene body or the extent of gene body methylation (area under the consensus peaks) between MT and WT CMML for the differentially expressed genes (Figure 1a). We further validated these results using methylation microarrays (Figure 1b). Among the MT CMML patients, there was a curvilinear relationship between gene body methylation and gene expression across all genes with intermediate gene body methylation being associated with the highest gene expression (Figure 1c). As an alternative unbiased approach, we identified 1595 differentially methylated regions (DMR, regions with FDR<0.05) in the validation data set. We mapped all hypermethylated regions to gene bodies requiring that there was no concurrent hypermethylation of the promoter region of the same gene. With this approach we identified one of the 707 up-regulated genes (0.14%) with evidence of isolated gene body hypermethylation (HBZ, log2-fold change in gene expression 4.25, FDR=0.0008, DMR area=1.61, DMR FDR=0.018). Representative examples of up-regulated mitotic kinases without evidence of differential gene body methylation are shown in Figure 1d (pie charts represent the mean β-values of microarray probes, p-values represent the comparisons of mean β-values between MT and WT CMML per gene).
Conclusions: Gene body methylation was positively associated with gene expression in MT CMML. However, the lack of differential gene body methylation between WT and MT CMML for the up-regulated genes make it an unlikely explanation for the observed increase in transcriptional activity among patients with MT CMML.
Ordog:Millipore Sigma: Patents & Royalties.
This icon denotes a clinically relevant abstract

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal